Proteins, Molecules, & Structural Biology
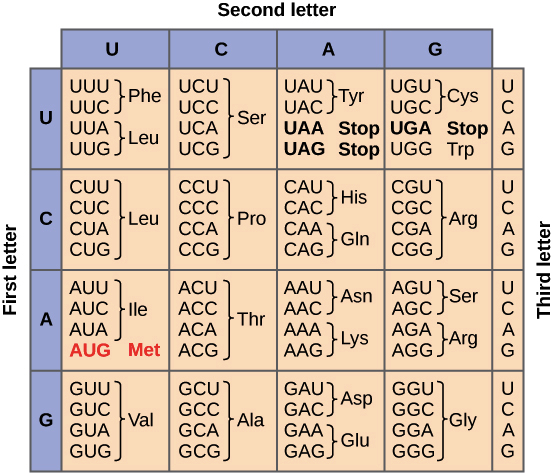
64 codons → 20 amino acids + stop signals (UGA, UAA, UAG). Always 5’ → 3’ end. Sequence constrains Structure. Structure is inextricably linked to function.
Apropos structure, see also: Intrinsically disordered proteins.
NLP analogues?
| Concept | Analogue |
|---|---|
| Token | Amino Acid |
| Word/n-gram | Motif (binding site) |
| Phrase | Domain (~50-300 AAs) |
| Grammar | 3D fold constraints |
| Corpus | Like UniProt (> 200M sequences) |
This is a pretty weak mapping though.
The generative process of language is not the generative process of proteins. But there are biochemical rules and grammer; the idea is that you can only take this so far (people certainly are gung-ho about tossing sequences into LLMs).
Protein Structure
How do you figure this out? You have crystallography, Cryo-Electron Microscopy (since 2015), NMR. But each has biases you should be aware of.
CASP is the protein structure prediction contest. “Here’s totally new proteins; predict structure.”
Data Gap
There are a lot of protein sequences (easy, read genome). But we don’t know much about the structure or function of proteins (vary across species). There’s PDB but it has ~250,000 which is 0.1% of total.
There’s also function labels (ClinVar, DMS/MaveDB).
There’s also AlphaFold but prediction truth. There are known areas where it is weaker (e.g. synthetic proteins — think about what it’s trained on (biological/natural sequences!)) and stronger. Proteins that lack an evolutionary signal negatively affect prediction tasks.
Evolution
Homologs → Orthologs (speciation; conserve function) + Paralogs (gene dupes; _may diverge in function)
Ancestral Gene -> Duplication -> Haemoglobin | --- Paralogs
-> Myoglobin |
Now Haemoglobin/Myo and in whale and human are orthologs.
Sequence Alignment
Pairwise: match, mismatch, gap. How you figure out if two proteins are homologs or Paralogs (or nothing). The foundation of classical protein analysis is aligning homologous sequences (Multiple Sequence Alignment).
BLOSUM62: Figure out the substitution frequencies. Needleman-Wunsch, Smith-Waterman, etc.
The Legend of Abraham Wald and survivorship bias…
You are looking for “positions 12 and 86” always change together… this may mean that there’s some 3D interaction happening. You can certainly use position-specific scores but they ignore this context of coevolution.
AlphaFold does very well when it doesn’t have evolutionary homologs; it’s modeling/capturing the evolutionary signal. It uses “MSA Transformers”.
There are all sorts of mutations (missense, nonsense, etc) but the key question is: is a missense mutation benign or pathogenic?
Designing Molecules to deal with Proteins
We’re dealing with small molecules. There’s the 2D skeletal representation, ball-and-stick, space-filling (shows shape), and… SMILES strings 😄
SMILES Strings → Sequences Models
Graph of Skeleton → GNN
3D Coordinates → Equivariant Models
Chirality is a problem if you just encode using a graph structure (e.g. R-Thalidomide will put you to sleep, D-Thalidomide will mess with your fetus (teratogenic)).
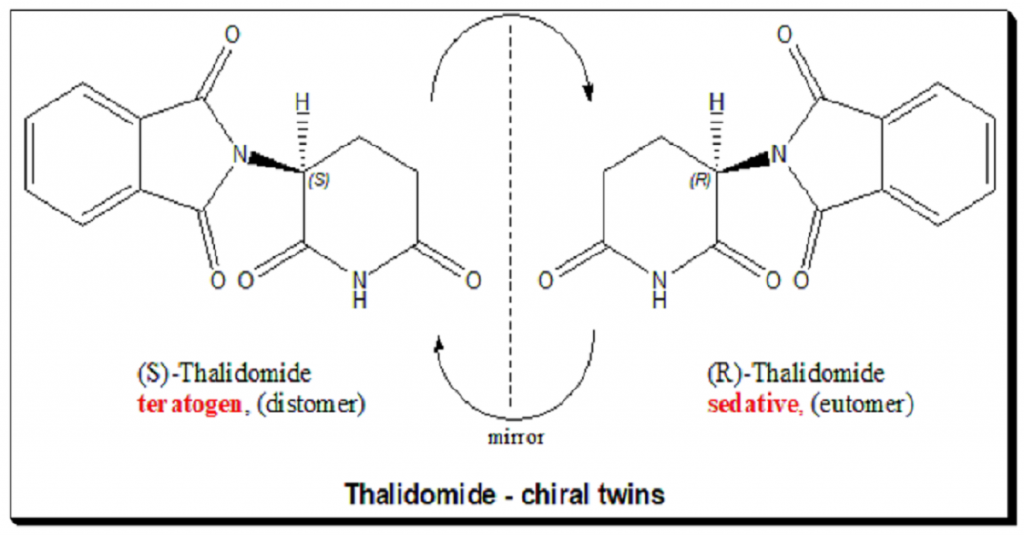
Enantiomers have the same 2D structure but different 3D handedness.
Now you can get Morgan/ECFP Fingerprints where you hash local substructures (like rings above) into binary vectors and do Tanimoto similarity. But you can have a high similarity but miss out on the potency.
The input representations that we have are not smooth but very jagged. If you want to change just one atom you may not be able to do this because of chem rules. You can sample very sparsely in molecular space!
In molecular modeling: split randomly and this leaks scaffolding (this will change… TODO?). You need to scaffold split — TODO read this. Your model will memorize that some substructures map to your label (e.g. teratogenic, etc.)
Molecular Docking
The question: Given a protein and its structure, how well will your drug ‘dock’ with it?
You can use physics models/molecular dynamics here and use top scoring ‘docking’ hits. Then hit the lab. Now that raw docking scores true affinity.
Drug Discovery
Target → Stucture → Hit finding → Lead finding → Preclinical Trials